$Revision: 1.26 $, covers Java TreeView 1.1.5
$Revision: 1.26 $, covers Java TreeView 1.1.5
Table of Contents
- READ THIS FIRST
- Acknowledgements
- 1. Installation and Introduction
- 2. Feature Reference
- 3. Troubleshooting
- Bibliography
List of Figures
- 1.1. Contents of Java TreeView Distribution Archive
- 2.1. Url Presets Dialog
- 2.2. Dendrogram Color Presets Dialog
- 2.3. Karyoscope Coordinates Presets Dialog
- 2.4. Url Settings Dialog
- 2.5. Dendrogram Font Settings Dialog
- 2.6. Dendrogram Pixel Settings Dialog
- 2.7. Karyoscope Settings Dialog
- 2.8. Karyoscope Averaging Settings Dialog
- 2.9. Dendrogram Export Dialog
- 2.10. Dendrogram Component Layout
- 2.11. Scatterplot Component Layout
- 2.12. Creating a Scatterplot
- 2.13. Karyoscope Component Layout
- 2.14. Alignment Component Layout
- 2.15. File Formats in Java TreeView
- 2.16. Screenshot of Generalized CDT file in Excel
- 2.17. Screenshot of a Tree File in Excel
- 3.1. Screenshot of Running Java TreeView from the Command Line with 800 Megs of RAM
List of Tables
This manual has three chapters. Each chapter has a purpose:
To assist the novice with installation.
To document all features
To provide troubleshooting assistance
New users should glance over the contents to see what the program has to offer, perhaps reading a section if it may be of interest. Familiar users may return if some errors arise, or if there is a feature they remember exists, but don't remember how to use.
This manual may be partially out of date. It has not been fully checked since 1.0.7, please email the mailing lists or the project admins on jtreeview.sf.net if you notice a section that appears to be out of date.
I would like to thank the members of the Botstein lab for valuable discussions and feedback, in particular Michael Shapira, John Murray, Maitreya Dunham, Barbera Dunn, Matt Brauer, Michal Ronen and my advisor David Botstein.
I would also like to thank the many other people both inside and outside of Stanford who have given me feedback, including Jason Lieb, Christian Rees, John Matese, Gavin Sherlock, Michiel Jan Laurens de Hoon and Stéphane Graziani.
Table of Contents
Installation of Java TreeView should be fairly straightforward on modern operating systems.
The following steps will help guide you through.
1) To get started, download a recent version of Java TreeView from the main web page, http://jtreeview.sourceforge.net.
Distributions
- Windows
TreeView-1.0.5-bin.zip
- Mac OS X
TreeView-1.0.5-osx.dmg
- Applet
TreeView-1.0.5-applet.tar.gz
- Generic Java
TreeView-1.0.5-bin.tar.gz
- Source
TreeView-1.0.5-source.tar.gz
- Mac OS 9
TreeView-1.0.0-bin.sit followed by TreeView-1.0.5-bin.tar.gz. You need to unpack both, and then overwrite the TreeView.jar file in the TreeView-1.0.0-bin.sit folder with the newer one. This will update the treeview program, but still allow you to double-click the treeview icon.
For most operating systems, the application is distributed as a .tar.gz file. For MacOS9, there is a special MacOS9 .sit file.
2) Unpack the .tar.gz file, if your browser has not already done so.
3) Doubleclick the TreeView.jar([1]) file to start, or the TreeViewLauncher file on MacOS9.
Known Issues
Stuffit version 7.5 for windows does not properly unpack tar.gz files. Use WinZip on the windows platform instead.
Note
If you run into problems at any time, check Chapter 3, Troubleshooting Troubleshooting for solutions

Once you have downloaded and unpacked the distribution, you should be confronted with a set of files similar to those listed in Figure 1.1, “Contents of Java TreeView Distribution Archive”. Before reading further, you should run the TreeView.jar program to make sure your installation works. At this point, you might want to view a sample cdt or pcl file to get the hang of how it works. You can grab one from the examples section of the website or skip ahead to the file formats section (Chapter 2, Feature Reference, the section called “File Formats”) if you aren't sure whether your file is properly formatted.
Java TreeView offers many views of the data which are linked together.
Views offered by LinkedView
- Dendrogram
Displays dendrogram, in style of original TreeView
- Scatterplot
Displays scatterplot of data values or per-gene statistics
- Karyoscope
Displays genome-ordering of data values, allows averaging
- Alignment
Displays aligned sequence data, typically from clustalw or a similar program
In all these views there are visual cues to show which genes are selected. Any operation which selects genes in one view, either due to genome ordering, hierarchical clustering, or per-gene statistics, selects the genes in all views. This is because the application only maintains one list of selected genes. This list of selected genes can also be used to create exported images and data files such as gene lists and sub-pcl files using the Export features, documented in chapter 2.
[1] ".jar" stands for Java ARchive, and is a standard format for java programs and libaries. Some jar files, such as the TreeView.jar files, are set up to be executable. Others, such as those located in the "lib" folder, are not.
Table of Contents
The amount of memory can be specified using the standard arguments to the jvm, i.e. for 500 MB, use java -jar -Xmx500m TreeView.jar
Be aware that for windows, you may need to use javaw instead of java.
The main program options have a short and long form. An example command line to open a specified file using the "linked view" style would be java -Xmx500m -jar TreeView.jar -r http://jtreeview.sourceforge.net/examples/DLim_color.cdt
Main Program options
- -r <file/url> (--resource=<file/url>)
File or url to load.
- -t <type> (--type=<type>)
Open file with the specified type of viewer. Valid values are "linked" for multiple linked view (by default only dendrogram opens on new files), "kmeans" for k-means, "classic" for the classic, dendrogram only, and "auto" to autodetect (default)
- -x <plugin> (--export=<plugin>
Export image from command line using the specified plugin instead of opening an interactive window. This argument requires that a resource to export is specified on the command line as well. Currently only the "Dendrogram" plugin is specified.
When export to dendrogram is indicated using "-x Dendrogram", main program arguments can be terminated with "--" and additional dendrogram plugin arguments can be specified. An example complete command line would be:
java -jar TreeView.jar -r ./spellman.cdt -x Dendrogram -- -o /tmp/spellman.png -s 10x1 -a 0 -c 1
Dendrogram export options
- -o <filename> (--output=<filename>)
(required) Filename to store image output in
- -f <imageFormat> (--=<format>)
(optional) set output format, either 'png', 'gif' or 'ps' (defaults to extension of output file)
- -s <widthxheight> (--scaling=<widthxheight>)
(optional) set pixel scaling as pixels for each array x pixels for each gene, or 1x10 for one pixel per array column and 10 pixels for each gene row, if you wanted to have gene names in the output
- -a <headerIndexes> (--arrayHeaders=<headerIndexes>)
(optional) Comma separated list of array annotations to include in output. Example: -a 0 to include the first row of array annotations in the cdt file. By default no annotations are included.
- -g <headerIndexes> (--geneHeaders=<headerIndexes>)
(optional) Comma separated list of gene annotations to include in output. Example: -g 0 to include the first column of array annotations in the cdt file. By default no annotations are included.
- -h <pixels> (--atrHeight=<pixels>)
(optional) Explicitly set the height of the array tree.
- -w <pixels> (--gtrWidth=<pixels>)
(optional) Explicitly set the width of the gene tree.
- -b (--below)
(optional) boolean argument, moves the array annotations below the array tree
- -c <value> (--contrast=<value>)
(optional) Explicitly set the contrast
- -l <value> (--logcenter=<center>)
(optional) Take log base 2(data value/<center>) before applying the contrast. Useful if dealing with raw count data.
Note: Setting the contrast or logcenter values will modify the .jtv file and have a persistent effect when the file is reopened in java treeview
For operating systems which have a command line, a file or url can be specified. The following example demonstrates how to load the example off the website from the command line: java -jar TreeView.jar -r http://jtreeview.sourceforge.net/examples/DLim_color.cdt
In Java TreeView, some effort was made so that when you set settings once, they stay set even if the application is closed and reopened. Such things as colors, zoom settings, url settings, etc. have settings which are set on a per-document level. There are also presets for these settings which are stored globally. A particular preset can be designated the default, and is used when opening new documents.
The actual Presets and Settings are covered in the section called “Presets” and the section called “Settings” respectively. This section simply tells you where the settings and presets are stored.
All document settings are stored in a document-specfic .jtv file. These settings always take precendence over the presets. If you want to reset a document so that it uses all presets, just delete the .jtv file. At some point, a menu option to do this might get added.
Currently, there are two program-wide settings: the presets and the most recently used list of files. Program-wide settings are stored in a global configuration file whose location is platform dependant. To be exact, the following code is run to determine what file to use:
private String globalConfigName()
{
String dir = System.getProperty("user.home");;
String fsep = System.getProperty("file.separator");;
String os = System.getProperty("os.name");;
String file;
if (os.indexOf("Mac") >= 0)
file = "JavaTreeView Config";
else if (fsep.equals("/"))
file = ".javaTreeViewXmlrc";
else if (fsep.equals("\\"))
file = "jtview.xml";
else
{
System.out.println("Could not determine sys type! using name jtview.cfg");
file = "jtview.xml";
}
return dir + fsep + file;
}
On unix, this resolves to a .javaTreeViewXmlrc file in your home directory. On OSX, it resolves to a JavaTreeView Config in your home directory. On PC, it resolves to a jtview.xml file, although I'm not sure where windows considers your home directory to be.
Presets allow you to store commonly used settings for url links, colors, and other settings. This allows you to quickly apply commonly used settings to different documents. Moreover, the default preset is the setting which a new file gets. Presets are not specific to any document, which means they can be edited when no files are loaded.

Url Presets are the only presets which apply to many views, since url linking is a view-independant process. Java TreeView stores the url presets for arrays and genes separately, thus there are two sets of url presets, gene url presets and array url presets, which are treated essentially the same.
Java TreeView supports linking to external databases. In a Dendrogram or Karyoscope, clicking on a gene will cause a corresponding database page to be loaded in an external browser. Dendrogram also supports linking by array. Exactly what database page gets loaded depends on the Url Settings. The exact mechanism for load depends on the operating system, and can be a source of url bugs (see Chapter 3).
URL Presets allow you to store presets for databases you may wish genes and arrays to link to. There are several default presets which come with Java TreeView. You can delete or modify all of them, as well as add additional presets.
There is a special preset which disables linking. You can set this as the default url preset to avoid spurious linking.

This set of presets allow you to configure preset color schemes for the dendrogram component. It is also referred to as just color presets in TreeView, since there's no other views which could have color settings.

Java TreeView stores settings in a per-file manner. Thus, it is not possible to edit settings unless a file is loaded. Additionally, LinkedView stores many settings in a per-view manner. Specifically, only the url settings are document-wide; the rest are only per-view. Thus, you could have multiple dendrograms with different zoom and color settings, and multiple Karyoscopes with different averaging, all with the same underlying document.

Url Settings allow you to select from one of the presets, or to directly edit the url string. There is a special substring of the url string, "HEADER", which is replaced by a partcular gene or url header which you select from the pulldown. The default is to either use the first column for a pcl, or the second column for a cdt. In the original Eisen layout, this is the YORF column.
There is also a checkbox which allows you to disable url linking entirely. Whether this box is checked initially is determined by the default url presets.
What exactly the url settings are used for depends on the view. Generally, clicking on a gene will cause the url for that gene to be loaded in an external browser.

Choose the font used to render gene or array names and annotation.
This fairly complicated dialog has three major parts. The first part allows you to set the pixel scaling for the global and zoom views. The second part allows you to set the contrast. The third part allows you to set the color settings for the dendroview.
The pixel scaling determines how tall and wide the boxes are in both the zoom and global views. Basically, the larger the pixel scaling, the bigger the box. If the pixel scaling is less than one, the rows are averaged. This can make your data look better, since missing values disappear.
The contrast is the expression value which corresponds to fully induced. Any values greater than this will appear to be the induced color, and values between this and zero will appear to be a color between the zero and up color. The contrast is similarly used to color repressed boxes.
The color part allows you to set the up, down, zero and missing colors. You can double-click the boxes to get a color selection dialog, click a preset to load a color, and load and store color sets to files.
In LinkedView, a dendrogram must be active in order for this option to appear on the settings menu. Any settings made only apply to the active dendrogram.

Coordinates may be parsed from the current file, if it is formatted properly, or parsed from an external file. The proper formatting is discussed in the File Formats section. The settings files provided with Java Treeview are nothing more than minimal PCL files, which contain no data but have annotation columns for chromosome, arm and position. The coordinates in an external files are matched up with the loci in the current file using either the first column, or if the first column has the header GID the second column. This gives the expected result when operating on PCL or CDT files, provided that the id column is unique.
Loci which do not have coordinates are not displayed. However, loci which have coordinates but not expression data associated with them do affect the extent of the chromosome displayed.
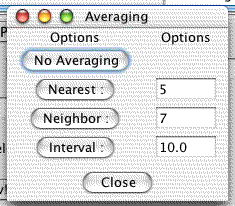
Allows one to average genes with their genomic neighbors using one of several algortihms.
Karyoscope Averaging Methods
- No Averaging
Do not perform any averaging
- Nearest
Average the nearest k genes. The nearest may all be on one side; this method strictly finds the nearest.
- Neighbor
Average the (k-1)/2 genes on the left and the (k-1)/2 genes on the right together with this one. If there are fewer than (k-1)/2 genes on a side, then fewer genes are averaged.
- Interval
Average genes over an interval of k units. The unit is the unit which the coordinates of the genes are specified in. This is generally base pairs for the supplied coordinates files.
Most displays support output to image formats such as PNG, PPM and JPEG. The dendrogram view also supports export to vector-based postcript files. Additionally, it is possible to export subsets of data to tab-delimitted text, i.e. gene lists and cdts.
Selected genes and data can be exported to tab delimited text using "Export->Export to Text File...". The export dialog allows you to select which annotation columns you would like to include, whether you want to include the expression data, and whether you want to include a header line. The default is to print out a simple gene list with no header.
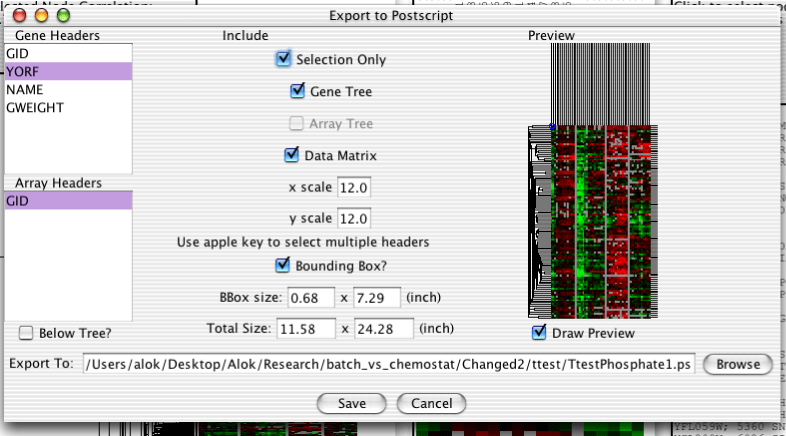
The dendrogram can be exported to either image files or editable Postscript for publication purposes. Postscript can be edited as objects in programs such as Adobe Illustrator, but can get prohibitively large with many genes and arrays. Image files are simply pixels, but can take up less space. To export to Postscript, select "Export->Export to Postscript" off the menubar. To export to images, select "Export->Export to Image". The following sections apply to both types of export. Although the above image is a screenshot of Postscript export, the only difference with image export is that there is no Bounding Box option. The purpose of the bounding box is discussed in the section called “Bounding Box”.
Both gene and array headers can be included in the exported image. The headers can be selected using the listboxes on the left side of the export dialog. Holding down the apple key on a mac, the Alt key on windows, or the Meta key on a unix machine will allow you to select multiple headers, or to deselect all headers.
Below the Listboxes is a checkbox which specifies whether the array names are to be output below the gene tree or not.
The top half of the column in the middle of the dialog contains checkboxes which control exactly what data in included in the exported image.
The "Selection Only" checkbox determines whether just the selected genes and arrays or all genes and arrays are output.
The "Gene Tree" and "Array Tree" checkboxes determine whether or not the gene and array trees should be included.
The "Data Matrix" checkbox specifies whether or not the actual data matrix should be included.
The bottom half of the middle column has a few text fields.
The "x scale" and "y scale" boxes set the size in pixels of the boxes in the data matrix.
The "Total Size" boxes contain the total predicted size of the exported image in inches. These fields are not used by the export machinery, and editing them has no effect.
A preview of the exported image using the current settings is displayed on the right side of the dialog. If the preview takes a long time to render for some reason, it can be disabled with the checkbox below.
The Bounding Box is a part of the PostScript standard which informs postscript renderers of non-standard page sizes. If you have problems seeing all of an exported postscript image then including the bounding box or making it bigger might help. Due to the vagaries of font rendering, there is no easy way for me to predict how long a particular text string is going to be. I take a decent guess, but sometimes the text will get clipped.
Corel Draw, Adobe Photoshop, and Adobe Illustrator all support the Bounding Box, and will correctly render exported postscript from Java TreeView.
Adobe Distiller ignores the Bounding Box and produces incorrect output on some platforms.
Java TreeView can be run in one of three modes which can be selected at the time time that a file is opened. Each window within a running instance of java treeview has it's own mode.
Java TreeView Modes
Auto
Classic TreeView
LinkedView
KmeansView
Auto is selected by default, and tries to do a good job of picking the best mode for a file. This should be sufficient for most users.
Classic TreeView is similar to the visualization program by Michael Eisen([Eisen 1998]). It consists only of a dendrogram view of the data.
LinkedView is a generalization of TreeView. The idea is to present multiple linked visualizations of the same data which is present in TreeView. Initially, it shows a dendrogram view, but a Karyoscope View, ScatterView, as well as a new Alignment View can be show if the appropriate data is available.
KmeansView is a specialized version of TreeView for viewing the output of Michiel de la Hoon's Cluster 3.0 K-means clustering ([De Hoon 2002] , http://bonsai.ims.u-tokyo.ac.jp/~mdehoon/software/) . This program is mostly similar to TreeView, although more k means-specific features could be added later.
The Dendrogram is one of three views which can be created in the LinkedView application. It is also the bulk of the TreeView application. The dendrogram has a lot of components, so I've gone ahead and given them names, so that the description of the features will not be confusing.
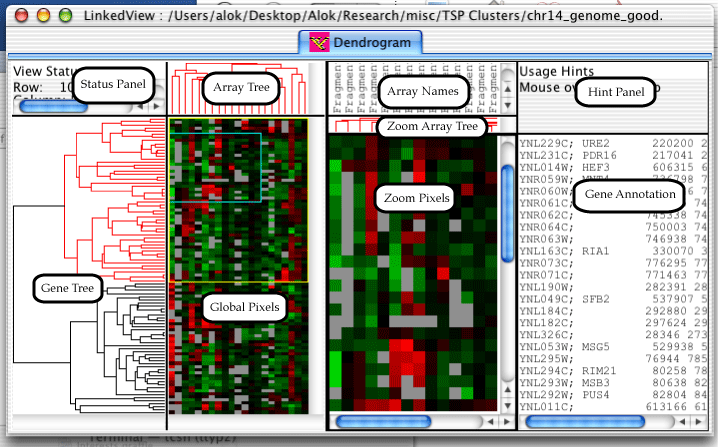
The status panel displays different information depending upon which component the mouse is over. The hint panel displays hints on how to use the component.
Genes can be selected in the dendrogram by either clicking and dragging on the global pixels or by clicking on a node in the gene or array trees. A zoomed in view of the selected genes will appear in the Zoom Pixels pane, a yellow rectangle will appear on the global view indicating which genes are selected, and a blue rectangle will appear inside the yellow rectangle indicating which genes are currently visible in the Zoom Pixels pane.
Holding down the shift key while dragging on the global pixels will cause the exact range of arrays to be selected; by default all arrays are selected. Once a range is selected, pressing the arrow keys moves the selected rectangle around. Holding down the control key while pressing the arrow keys will grow and shrink the selected rectangle.
Clicking on a node in the array or gene trees will select all descendants of the node. The selected node and descendants will be colored in red. At this point, pressing the arrow keys will change which genes are selected. Up will select the parent of the current node, left and right will select the left and right children, and down will select the child with more descendants.
Provided the url link settings (described in the section called “Url Settings”) are set appropriately, clicking on a gene annotation or an array name will cause a browser window to open with details on the gene or array.
To add color to gene names, you must add a column named "FGCOLOR" before the GWEIGHT column. Don't have a GWEIGHT column? Well, you need to add that immediately before your first array column. For each gene, you need to specify a the section called “ Hex Codes for Name Coloring ” for the color you want that gene's name to appear in. Clearly, it is a good idea to write some kind of excel function which computes it for you, or a perl script or something.
To add color to array names, you must add a row named "FGCOLOR" before the EWEIGHT row. If you don't have an "EWEIGHT" row, you must it immediately before your first row of gene expression data. For each column in the EWEIGHT row, you must specify a the section called “ Hex Codes for Name Coloring ” for the color you want that array's name to appear in.
Hex color codes are commonly used in html documents to specify colors. The format is as follows: #RRGGBB. The RR value is a hex number between 00 and FF which specifies how much red you want. In other words,
- #FF0000 is pure, bright red
- #00FF00 is pure, bright green
- #0000FF is pure, bright blue
You can of course mix values, and use lower values than FF for less intense colors. you can either find colors by trial and error, or do a quick web search for hex color codes. It is recommended that you edit the cdt file in excel or write a perl script to add the color codes easily.
The Scatterplot is one of three views which can be created in the LinkedView application.
The scatterplot is a fairly simple component. Clicking and dragging will select the genes within the rectangle. Holding down Control will allow you to select multiple disjoint sets of genes. Moving the mouse around will tell you the X and Y coordinates of the mouse at any given point.
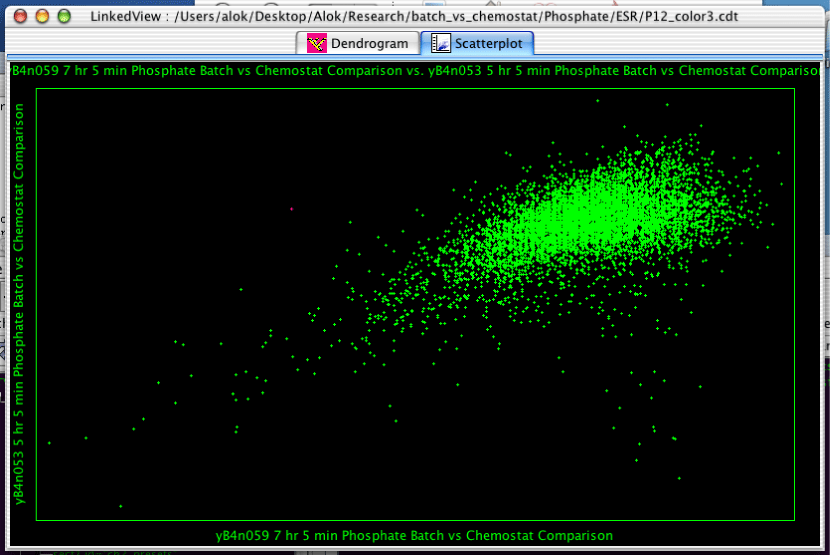
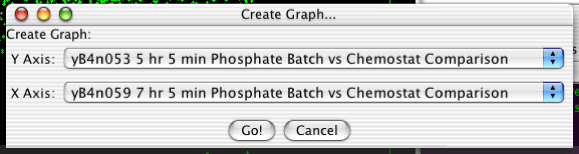
To create a scatterplot, select "Analysis->Create Scatterplot..." from the menubar. You will then be confronted with the dialog in Figure 2.12, “Creating a Scatterplot”. The two pulldown menus determine what will be plotted on each axis. There options include the various arrays, the actual row number of the gene (called INDEX), and any annotation columns.

The Karyoscope is one of three views which can be created in the LinkedView application. It can be created by selecting Analysis->make Karyoscope in LinkedView. There are three major regions to the screen in the Karyoscope. The upper left panel is the parameter panel, the upper right panel is the info panel, and then lower part of the screen is the karyoscope proper.
There are several settings which apply to the Karyoscope which have been previously covered, including the Coordinates the section called “Karyoscope Coordinates Settings” and Averaging the section called “Karyoscope Averaging Settings” and Url the section called “Url Settings” Settings. The remaining features are covered here.
Moving the mouse around the screen in the Karyoscope will cause information for the nearest gene to be displayed in the info panel. There will be a bright yellow line connecting the cursor to the nearset gene, so you know which one the info applies to. Clicking will activate the url link associated with the gene.
The layout of the genes in the Karyoscope is determined by the Coordinates Settings and the Parameter Panel. The coordinates settings specify the chromosome, arm and position of each gene. The Parameter panel specifies exactly how to translate this information into an on-screen location.
The Pixels Per Map specifies how many pixels each position unit stands for. Making this number larger will stretch out the chromosomes.
The Pixels Per Value specifies how many pixels each value unit from the data matix gets. Making this number larger will make the bars higher in the karyoview.
The width and height specify the actual width and height of the canvas on which the karyoscope will be drawn. Making these values larger will space out the chromosomes without actually changing the spacing of the bars or their height.
Since setting these values manually is tedious, there are two additional ways to navigate, which are preferred for general use. Clicking the "Auto" button in the parameter panel will set the width and height to match the available screen real estate, and then scale the pixels per map and pixels per value appropriately. Selecting a rectanglar region in the Karyoscope causes the screen to zoom in on that location. Technically speaking, it causes the selected region to exactly fill the screen, increasing the pixel per map and pixels per value proportionately.
Although there is currently no way to select genes in the Karyoscope, genes which have selected in other views are visibly marked. The type of marking is determined by the settings in the Parameter Panel. You can choose the type and size in pixels of the highlighting from the pulldown menus to the right of "Highlight Selectd with:".
Karyoscope Selection Highlights
- None
Do not highlight selected genes.
- Circle
Highlight selected genes with a solid circle.
- Disc
Highlight selected genes with a 1-pixel thick disk
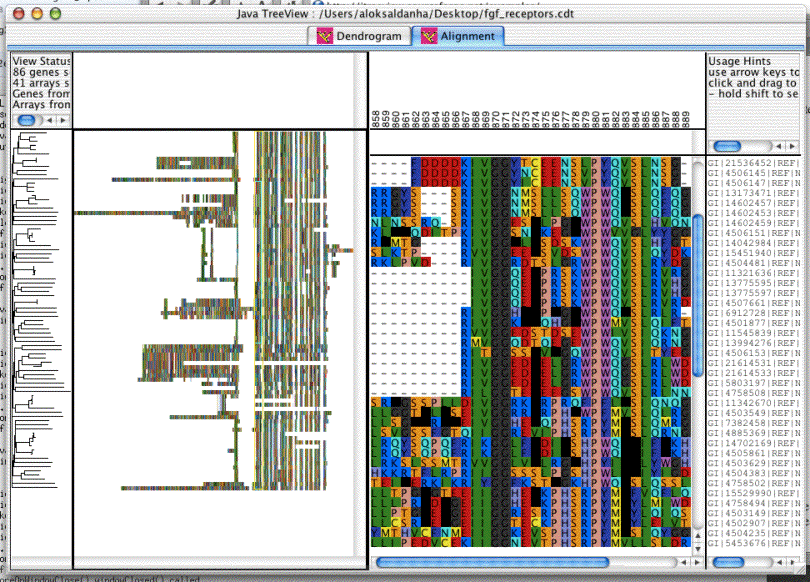
The Alignment view is very good for browing large alignments, even if you don't have any expression data for them. There is a utility, "aln2cdt.pl" available in the helper-scripts package from website to facilitate the viewing of clustalw alignments.
The alignment view relies up the existence of a column named "ALN" containing all the sequence data, with IUPAC symbols for the amino acids, and non-symbol spacer characters such as dashes designating the gaps. The alignment view will render the matching sequence into a dendrogram-view like two-level display, complete with gene tree.
There are two PERL scripts in the helper-scripts package available from the website which should aid in the usage of alignment view, aln2cdt.pl which will create a cdt with the appropriate columns, and potentially a gtr fie if there is a dnd file available, and appendPCL.pl, which will allow you to append expression data in the pcl format to the cdt alignment file.
For a detailed howto to assist in making your own alignments, please see the fgf receptor example on the website (http://jtreeview.sourceforge.net)

Java TreeView uses two file formats to represent data, both of which are tab-delimited text. The third file format is an xml formatted file which holds settings information for persistence (see the section called “Persistence”).
Java TreeView File Formats
Generalized CDT File (.cdt and .pcl)
Tree File (.atr and .gtr)
XML Settings File (.jtv and global settings file, see the section called “Global Settings”)
The use of tab-delimitted text makes these files easy to edit in spreadsheet programs such as Excel, as well as manipulate with other programs.
In order to view data in Java Treeview, there must be a Generalized CDT file. All other files are optional. The minimal GCDT file has a header row which contains the name of the unique id column, the name of the annotation column, and the names of the experiments, followed by one or more rows of per-gene data. Such a file can be created in Excel, and then saved as tab-delimited text.
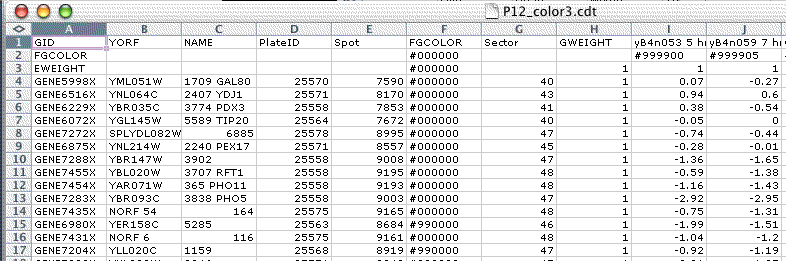
The generalized CDT file is a straightforward generalization of the CDT and PCL file formats. In addition to expression data, this file can contain additional per-gene and per-array annotation in columns before the GWEIGHT column or in rows before the EWEIGHT row. For backwards compatibility, if the GWEIGHT column is missing Java TreeView assumes the data starts on the third column, or the fourth column if the first column has the header GID. Similarly, if the EWEIGHT row is missing Java TreeView assumes the data starts on the second row. As a general practice, it is a good idea to include the GWEIGHT column and EWEIGHT row.
In addition, Java TreeView does special things with the first two or three columns. If the first column is GID, the second and third are assumed to be the unique ID and NAME columns. If the first column is anything other than GID, the first and second columns are assume to be the unique ID and NAME columns. The unique ID is used for gene list export, and for some matching purposes when necessary. The NAME column is displayed as per-gene annotation in the dendrogram and other views.
There are annotation column names with special meaning to Java TreeView, and are used for coordinates or to set the color of gene names. These special columns are described after the basic file format, and should be avoided as annotation names unless you want that specific behavior.
A generalized CDT file is a tab-delimitted text file with the following specifications. The leftmost column and topmost row are reserved for headers. The file must contain at least two columns followed by a column with the header GWEIGHT, and at least one row followed by a row with the header EWEIGHT. Any rows and columns before the EWEIGHT and GWEIGHT are treated as annotation, and any after are treated as data. If a data value is missing or cannot be converted into a number, it is treated as not found. The annotation is kept in string form, and parsed by views as appropriate.
Some headers have special meaning to particular displays. This is a catalog of headers and their meaning to particular displays.
Table 2.1. CDT Column Headers With Special Meaning
| Header | Display | Meaning |
|---|---|---|
| FGCOLOR | Dendrogram | Color in which to render text for particular gene |
| BGCOLOR | Dendrogram | Color in which to render background for particular gene |
| LEAF | Dendrogram | The TIME at which this branch should be terminated. Used to indicate apopotosis in cell lineages, as well as phyogenetic distance in sequence alignments. See also TIME header for tree files. |
| CHROMOSOME | Karyoscope | The chromosome on which the gene is located, a natural number |
| ARM | Karyoscope | The arm of the chromosome, either "L", "R", or "1" meaning left, "2" meaning right. |
| POSITION | Karyoscope | The distance of the spot from the centromere in arbitrary units |
| GROUP | Dendrogram | Defines a partitioning of genes. The current (1.0.13) implementation of Dendrogram will insert a gap every time the GROUP value changes. At some point in the future, I may make it so that all genes with identical GROUP values are put in one cluster. |
Table 2.2. CDT Row Headers With Special Meaning
| Header | Display | Meaning |
|---|---|---|
| FGCOLOR | Dendrogram | Color in which to render text of array name |
| BGCOLOR | Dendrogram | Color in which to render background of array name |
| GROUP | Dendrogram | Defines a partitioning of arrays. The current (1.0.13) implementation of Dendrogram will insert a gap every time the GROUP value changes. At some point in the future, I may make it so that all rows with identical GROUP values are put in one cluster. |
In order for Karyoscope to correctly display gene expression data by chromosome location, it needs to know where exactly to position each unique ID. To this end, it looks for annotation columns with the names "CHROMOSOME", "ARM" and "POSITION", which designate the chromosome, arm and position of a particular gene. "CHROMOSOME" should be a natural number indicating which chromosome the unique ID is on, "ARM" should be either "R" or "L" indicating the arm, and "POSITION" should be a real number indicating how far from the centromere the unique ID is. There is really no restriction on the units for position; bp or kb are natural choices.
A coordinates file is simply a generalized CDT file which has such columns. The coordinates files supplied with Java TreeView do not contain any expression data; they consist entirely of the unique id column, the chromosome, arm and position columns, and the required GWEIGHT column. However, any other generalized CDT file with the correct columns can serve as a coordinates file.
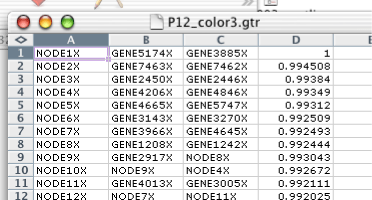
Traditionally, tree files have no header, and consist of four columns. Each row represents a node in either a gene tree, for the GTR file, or an array tree in the ATR file. For each row, the first column is the identifier of the node, the second column is the left child of the node, the third column is the right child, and the fourth column is the correlation between the left and right child. This fourth column is used by Java TreeView to determine the height of the node when rendering a tree.
By analogy to the CDT file, the tree files have been generalized in Java Treeview. Generalized tree files have a header line identifying the different columns. All generalized GTR/ATR files must have NODEID as the name of the first column. Tree files with any other string in the first row of the first column will be treated as legacy tree files. All of the rows will be treated as defining nodes, and the headers will be assigned the headers "NODEID", "LEFT", "RIGHT" and "CORRELATION". The meaning of these headers, and others, are described in the section called “ Tree File Headers ”.
All tree file headers can be displayed as node annotation in the dendrogram view. After loading a file in java treeview, select "Settings->Annotations..." to open the annotations dialog. Click "Gene Nodes" or "Array Nodes" and then select the desired headers. If you then mouse over the tree, the annotation for the select node will appear in the info pane.
Headers that have meaning beyond being used as annotation are described here.
Table 2.3. GTR/ATR Column Headers With Special Meaning
| Header | Display | Meaning |
|---|---|---|
| NODEID | Dendrogram | The value in this column serves as the identifier for the node. It must be unique. |
| LEFT | Dendrogram | The NODEID of the left child of this node. If the left child is not an internal node but a gene from the CDT file, the value should be the gene identifer, i.e. the value in the first column of the CDT file. |
| RIGHT | Dendrogram | The NODEID of the left child of this node, with leaf nodes handled as for the LEFT column. |
| CORRELATION | Dendrogram | The correlation value for this node. If this column appears, the implication is that the nodes in the tree should be arranged with a value of -1 most distal from the data matrix, with a value of 1 immediately proximal. This column and the TIME column should not appear in the same tree file. |
| TIME | Dendrogram | The time at which this node branched. Java Treeview is used by some to display cell lineages. For this purpose, it is useful to arrange branches in the tree by time at which the cell divided. This column is also used for indicating branching in sequence alignments, although TIME means phyogenetic divergence time in that context. If this column appears, the implication is that the nodes in the tree should be arrange with a value of 0 most distal from the data matrix, and that the node with maximum time value should be immediately adjacent to the data. |
| NODECOLOR | Dendrogram | The color value for this node. In the abscence of this column, the dendrogram is rendered black when not selected. Even when this column appears, selected nodes are rendered red. |
Some users have reported problems unzipping the distribution. The problems encountered range from all files getting dumped in the root directory to not being able to unzip the distribution at all. This stems from my approach of using a single, relatively platform independant format, the .tar.gz format. The solution is to just use an unzipping program which handles .tar.gz properly.
Compatible Unzipping Programs
- GNU tar/ GNU gunzip
These are the command line tools I use to create the distribution. They are ubiquitously available on all unix and unix-like operating systems, including Mac OS X, if you know where to look.
- Winzip
This is the recommended unzipper on the windows platform. I have heard reports that it doesn't do the right thing if you simply double-click the .tar.gz file. Instead, you may need to right-click, and select the "Expand to folder TreeView-blah" option.
- Stuffit on Mac OS 9
This is the recommended unzipper for Mac OS 9. This should definately work on the specially packaged OS 9 distribution, which comes as a .sit file.
- Stuffit on Mac OS X
As far I know, this should work fine. Just make sure you download the correct (.tar.gz) distribution of treeview.
Incompatible Unzipping Programs
- Stuffit on Windows
This is known not to work in at least some cases. If it does not make a directory structure like Figure 1.1, “Contents of Java TreeView Distribution Archive”, then just go get winzip.
The most common problem I have seen is that java is not installed on the system. The symptom is that nothing happens, or something confusing happens, when you doubleclick the launcher. The solution is to install a recent version of the java runtime environment. For Windows or Unix systems, this involves a quick download from the sun website,
Mac OSX comes with java standard. For Mac OS9, you may need to get the Mac OS Runtime for Java (MRJ) from the apple website,
Clicking on a gene in the dendrogram and karyoscope views is supposed to open a web browser window to a relevant database entry. Unfortunately, there is no standard way to do this in windows. When the linking is bad, typically clicking a gene name launches a DOS terminal window where the window's title equivalent to the URL, but no browser window comes up.
I have tried several things, but nothing works for all versions, and if you are not lucky you will be using one for which the current incarnation does not. Luckily it is easily fixed. Go to start->execute.. and entry "cmd" to get a command line shell. Try the following variants to see how your version of windows likes to do linking:
start http://www.google.com
cmd /c start http://www.google.com
start "http://www.google.com"
cmd /c start "http://www.google.com"
By default, #2 is used in version 1.0.5. If you require something else, you can enter the entire command as the URL string in java treeview, i.e.
start http://genome-www4.stanford.edu/cgi-bin/SGD/locus.pl?locus=HEADER
Including the quotes should guard against forbidden characters in the HEADER, as explained in my code:
// The problem with the above is that special shell characters, notably & | ( ) < > ^ ,
// need to be escape, or put in double quotes (which can be doubled to escape them,
// but I don't think that's necessary.)
When OSX tries to open a bad url, sometimes something breaks in the native code. If you run the application from the command line, you will see errors like the following:
2003-03-18 21:41:23.197 java[3492] LSOpenCFURLRef() returned -50 for URL (null) in OsX
This output indicates that an underlying C function failed. The java thread running this just dies without throwing an exception, so there's no way for me to detect it. The symptoms on the user level is that clicking has no effect. If you think this might be the case, double check your Url settings, in particular the preview, and make sure that the url is well formed.
Problem: I want to display things that are similar by Genename. This fucntion is present in your program when you select genes that have the same annotation(Analysis->Find...) , and then show a summary of these genes (Analysis->Summary Window...). Is there any way to save an image from the summary window?
Solution: There may be a way in the future to directly export from the summary window. However, in the meantime you can export data for the selected genes to form a valid CDT file, which can then be opened with Java Treeview and used to form images, etc.
First, Select the genes using the annotation search as you described. Then, use Export->Data... to export the data just for the selected genes. If you accept all the defaults, it will make a well-formed CDT file that you can then reopen and export to an image.
If you output a giant Postscript file and then try to open with adobe illustrator and get the error "The MPS parser is unable to parse the file" it most likely means that the postscript file created by java treeview requires a bigger canvas to display than the maximum canvas size of illustrator. I don't have a good answer on how to fix this, other than lowering the xscale and yscale.
Symptom: Trying to export some big clusters with names and dendogram to GIF, but continually getting an 'Out of memory' error. Able to export results only if you scale down the cluster image (i.e. using low X & Y scale values).
Solution: The gif encoder in java treeview sucks. Use Export->Export to bitmap... (ppm) format and then convert to gif with some software with a better gif encoder. "GraphicConverter" which comes by default with Mac OS X works well. There are also various windows options, such as "Image Transformer", which is shareware.
Note- this section is deprecated, as the GIF encoder is no longer used.
Running out of memory is tough to detect and deal with in java, since most of memory allocation is hidden from the java developer. If you are working with large data sets, or on a computer with a small amount of RAM, it is easy to run out of memory because java is not very memory efficient, and more importantly most Java Virtual Machines (JVMs) are too stupid to grow to take up more memory than some default arbitrary maximum.
There are only two possible solutions to the out of memory problem in java. Either I write a platform-specific launcher for each platform that informs the JVM of the extra ram, or you run the application from the command line and tell the JVM about the RAM yourself. Currently, only the latter is supported.
To run the application from the command line, first get to a terminal. This should be obvious to someone on a unix machine. On Mac OS X, run the Terminal program which is in the utilities folder. On Windows, select Start->run... and then type cmd <return> . Curously, on Mac OS 9, this problem does not seem to arise. I suspect this is because the OS 9 JVM is smart enough to use the per-file memory settings.
Next, switch to the directory containing the java treeview distribution (using either cd or chdir or whatever), and then type
java -Xmx###m -jar TreeViewLauncher.jar
Where the number following Xmx is the maximum amount of memory in megabytes that the JVM is to use. You may as well specify the available system RAM; the JVM won't take it unless it needs it. Also, make sure that you're in the directory with the jar files so that the command will work. Check out Figure 3.1, “Screenshot of Running Java TreeView from the Command Line with 800 Megs of RAM” for an example.

Question: I have two columns before I get to my data, the first being ID(containing accesion number), and the second is "NAME" (containing Unigene ID). The data follows in each column after these two. When treeview loads it up, it only displays the 2nd column "NAME", instead of both columns data when highlighting aspects of the cluster. Is this right?
Answer: Yes. TreeView used to display all annotations, but it annoyed some people. To specify the ones you want, use Settings->Annotations...
On the windows and mac platforms, the system default browser is used to open urls. In order to change it, you must muck around with your control panel.
On unix, there is no notion of a system default web browser, so I just hardcoded in a call to netscape. If you don't have netscape, or you want to use another browser, just alias netscape to that other browser from the shell in which you're launching java treeview. Something like the following should work:
alias netscape mosaic
java -jar TreeViewLauncher.jar
If the above doesn't work, then the JVM is spawning a sub-shell to run the call to netscape in. In that case, you need to add the alias to your .profile or equivalent, or perhaps just add a link to your preferred browser such as the following:
ln -s /usr/bin/mosaic netscape
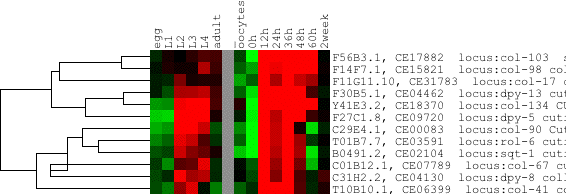
Some people have been thrown off guard by dendrograms such as the one depicted above. The strange appearance of the tree, where the parent node of dpy-8 and col-67 has a lower correlation than its parent, is a correct representation of the clustering algorithm used to generate the tree. Indeed, the correlation between the two genes is .889, whereas the correlation of the next join up is .908. This simply means that the average of dpy-8 and col-67 is more similar to the next node up than they are to each other (and, by implication, than either dpy-8 or col-67 alone are to the next node up).
If such behavior disturbs you, please use a different correlation metric, such as complete linkage. Certain other visualization programs, such as the original Treeview, artificially force parent nodes to have a correlation equal to or less than that of their children, but I choose to accurately represent the information in the GTR and ATR files.
Typical symptoms are the application seems to lock up after a fairly innocuous user action, and then the application suddenly quits. If you ware running from the command line, you may see a report of a "bus error" or "segmentation fault". Running "Console", an application that comes with OSX and can be found in the Utilities subfolder of the Applications folder, should reveal something like
May 18 11:27:19 Alok-Saldanhas-Computer /Users/aloksaldanha/Desktop/code/java/LinkedView/bundle/TreeView/Java TreeView.app/Contents/MacOS/JavaApplicationStub: An error report file has been saved as /Users/aloksaldanha/Library/Logs/JavaNativeCrash_pid2964.log. Please refer to the file for further information. May 18 11:27:21 Alok-Saldanhas-Computer crashdump: Crash report written to: /Users/aloksaldanha/Library/Logs/CrashReporter/JavaApplicationStub.crash.log May 18 11:28:03 Alok-Saldanhas-Computer WindowServer[188]: Reserved range exhausted. (0xbbf4b000 to 0xbc1d1000 goes out of bounds) May 18 11:28:10 Alok-Saldanhas-Computer /usr/bin/java: An error report file has been saved as /Users/aloksaldanha/Library/Logs/JavaNativeCrash_pid2971.log. Please refer to the file for further information.
as opposed to
[error] apple.awt.EventQueueExceptionHandler Caught Throwable : [error] java.lang.ArrayIndexOutOfBoundsException: Array index out of range: 52133 [error] at org.gjt.sp.jedit.Buffer.getLineOfOffset(Unknown Source) [error] at xml.TagHighlight.updateHighlight(Unknown Source) [error] at xml.TagHighlight.access$000(Unknown Source) [error] at xml.TagHighlight$1.actionPerformed(Unknown Source) [error] at javax.swing.Timer.fireActionPerformed(Timer.java:271)
In general, if Java Treeview crashes it is advisable to check the console for messages. The former indicates a bug in the JVM, not in Java Treeview, which at some point I should write up as a formal bug report. Something like the latter indicates a bug in Java Treeview, although this particular instance was another java program. There are at least two causes for the JVM crashes, one of which is somewhat understood. If you are launching Java Treeview by double-clicking a jar file, and you have multiple views open, you could be triggering a bug in the Mac OSX Java libaries related to font rendering. Either run Java Treeview with the following command line:
java -Xmx500m -Dapple.awt.TextAntialiasing=false Treeview.jar
or just use the double-clickable application that comes in the .dmg download.
Java Treeview may also crash when you select entries from the most recent file listing. It is not clear what causes this or how to fix it at the moment.
[Eisen 1998] Proceedings of the National Academy of Science, U S A. 1998 Dec 8. 25. 14863-8. “Cluster analysis and display of genome-wide expression patterns”.