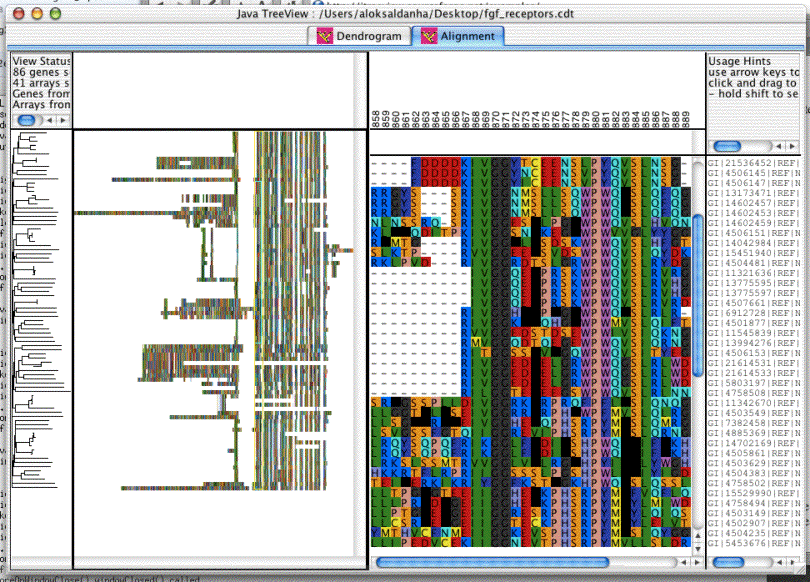
The Alignment view is very good for browing large alignments, even if you don't have any expression data for them. There is a utility, "aln2cdt.pl" available in the helper-scripts package from website to facilitate the viewing of clustalw alignments.
The alignment view relies up the existence of a column named "ALN" containing all the sequence data, with IUPAC symbols for the amino acids, and non-symbol spacer characters such as dashes designating the gaps. The alignment view will render the matching sequence into a dendrogram-view like two-level display, complete with gene tree.
There are two PERL scripts in the helper-scripts package available from the website which should aid in the usage of alignment view, aln2cdt.pl which will create a cdt with the appropriate columns, and potentially a gtr fie if there is a dnd file available, and appendPCL.pl, which will allow you to append expression data in the pcl format to the cdt alignment file.
For a detailed howto to assist in making your own alignments, please see the fgf receptor example on the website (http://jtreeview.sourceforge.net)